 COVID-19 Docking Server
COVID-19 Docking Server
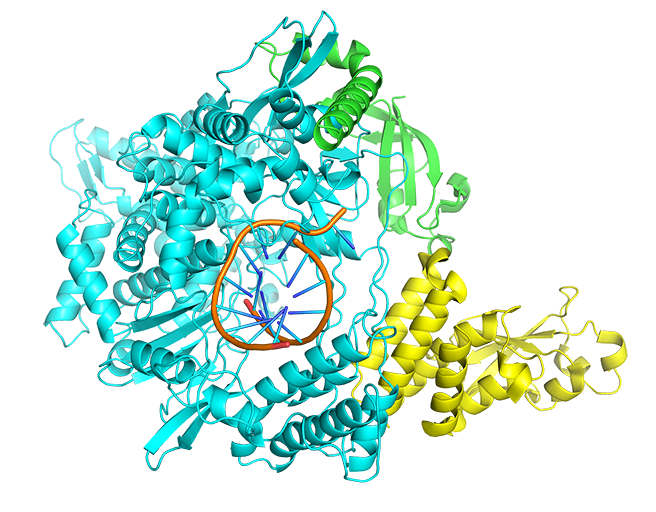
A web server for docking small molecule, peptide or antibody to COVID-19 protein targets.
Submit | Check Result | Job Status | Result Example | Target Annotation | Tutorial | Citations
|
COVID-19 Docking Server
A web server for docking small molecule, peptide or antibody to COVID-19 protein targets. Submit | Check Result | Job Status | Result Example | Target Annotation | Tutorial | Citations |
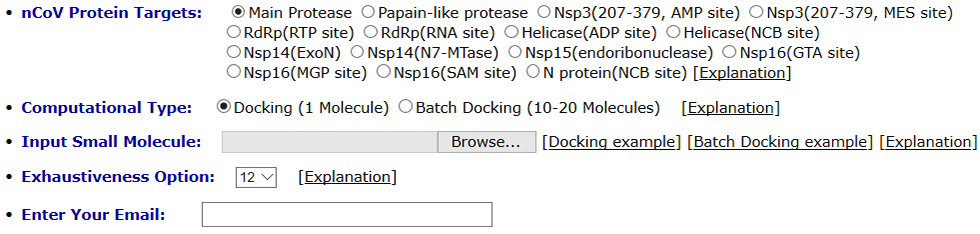
You need to choose one of the COVID-19 targets and upload small molecules in strict smi, mol2 and sdf formats. Open Babel (License: Open Babel Official Page) is used for format transformation or 3D coordinate generation for the uploaded files. Autodock Vina (License: AutoDock Vina Official Page) is used as docking engine. You should choose the computational modules before you upload the small molecule files: if “Docking” is selected, only one small molecule should be uploaded, and top 10 binding modes will be displayed on the result page. If “Batch Docking” is selected, 10-20 molecules should be uploaded, and the top 10 molecules ranked by the scoring function will be displayed on the result page. If you have 1-10 molecules need to be evaluated, then you have to upload the molecules one by one and choose the “Docking” module. Exhaustiveness is a parameter of the docking search. Increasing this value will increase the time linearly and decrease the probability of not finding the minimum exponentially. An email will be sent to your email address, containing a directed link to access you docking results.
After your job completes, you could enter the job ID to access the result page.
Alternatively, you can access the docking results by the corresponding link in your email. The download url is provided as: http://ncov.schanglab.org.cn/download.php?dir=*************
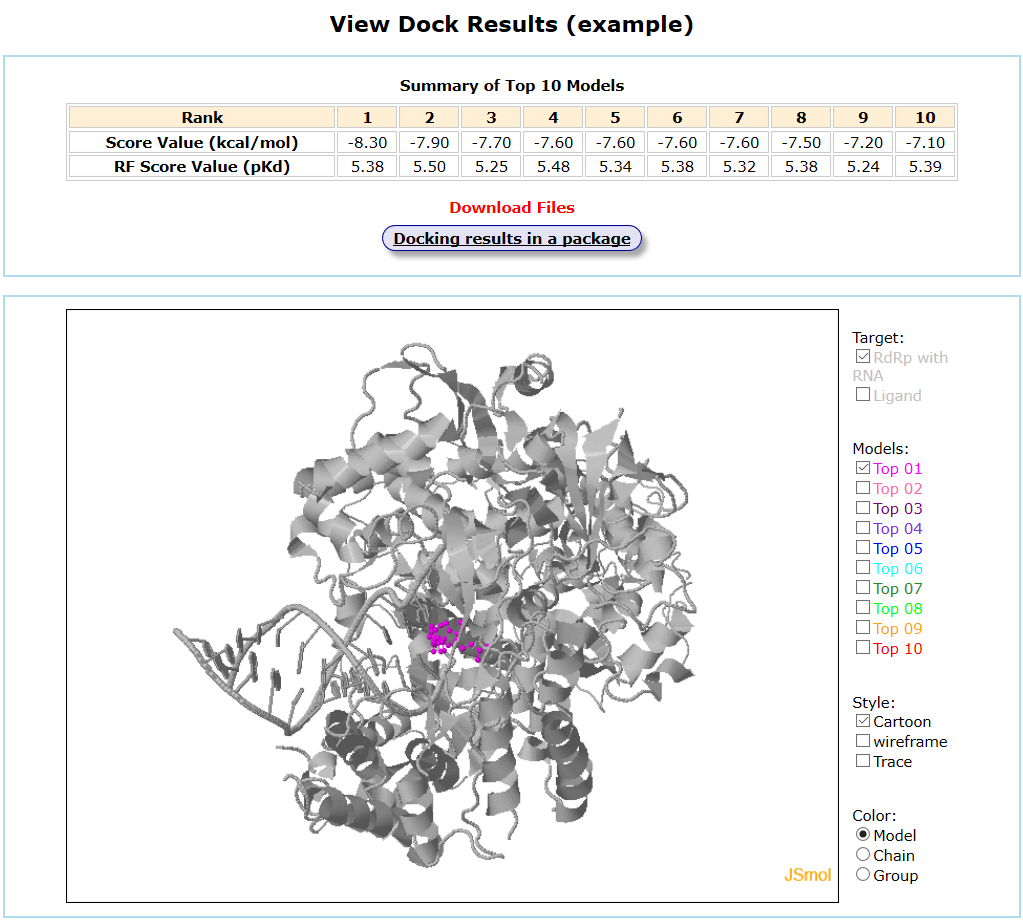
When the job is completed, the server will send an email to the user. By default, the server only displays the top 10 models. The selected COVID-19 protein target is renamed Input_R.pdb. The top 10 models are named TopL1.pdb-TopL10.pdb, respectively. The models can be visualized in 3D by JSmol (License: Jmol Official Page). The scored file is named as Score.dat. For small molecule docking, ligand poses docked against a receptor by AutoDock Vina are re-scored by RF-Score-v4 (License: RF-Score-v4 software Page). The re-scored file is named as Score_pKd.dat. You can view and download your docking results from the result page.
You need to choose the COVID-19 protein target and upload ligand (peptide or antibody) proteins in strict pdb format. The PDB checker, such as Molprobity, should be used to check the file and fix any potential errors in pdb file before you upload it. For peptide and antibody docking, CoDockPP (CoDockPP Server) is used as docking engine.
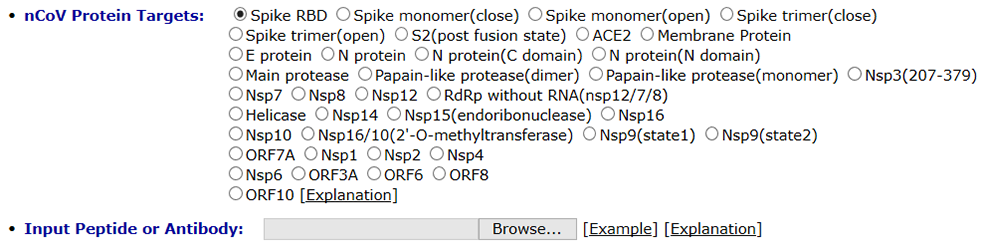
The protein docking server can perform global docking and site-specific docking to predict the binding mode between two proteins. You can enter one constraint residue on the receptor interface and another one on the ligand interface.
The binding site residues are provided in the text box as follows
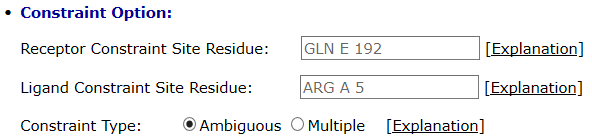
The example shows that GLN192 in chain E of the receptor and ARG5 in chain A of ligand are chosen for site constraints. User could enter one residue into either the receptor box or the ligand box, or he could enter one residue into receptor box and one into the ligand box simultaneously. If only one residue on the ligand or receptor is defined, then the conformations with the specific residue on the interface of the complex are retained. When the user defines one residue on the receptor and the other one on the ligand simultaneously, then he needs to choose the constraint type: ambiguous constraint or multiple constraints. When the ambiguous constraint is selected, the conformations are retained with at least one selected residue on the interface. When the multiple constraints are selected, the conformations are retained with both of the residues on the interface.
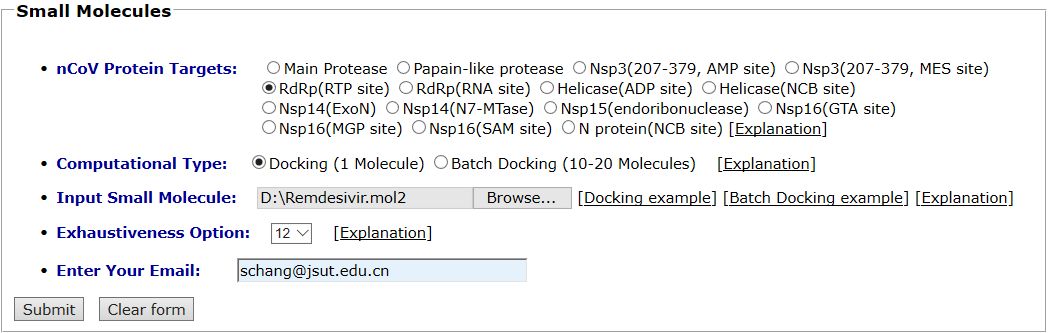
RNA-dependent RNA polymerase (RdRp) and its inhibitor Remdesivir are taken as an example for small molecule docking. RNA-dependent RNA polymerase is selected as the protein target. The “Docking” mode is selected and the mol2 format of its inhibitor Remdesivir in PDB 7BV2 is uploaded as the input ligand. Exhaustiveness option is 12. The input interface is shown as follows.
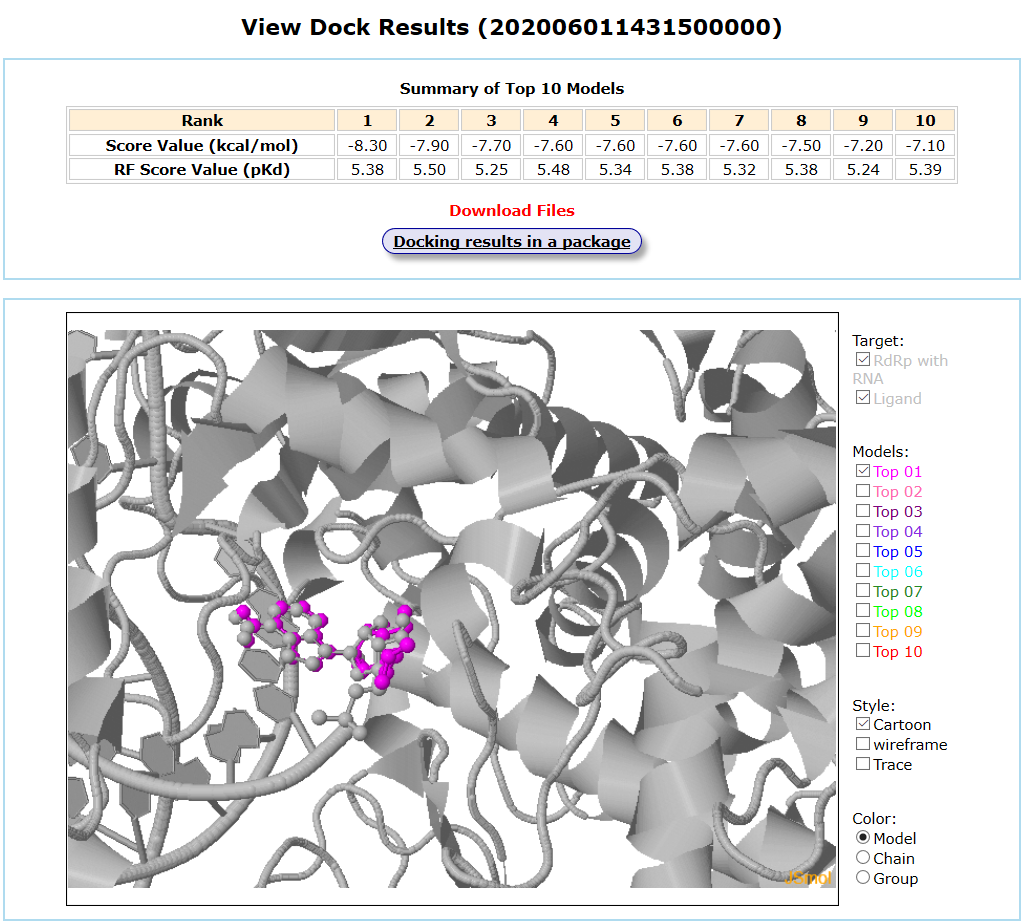
Results are available on the page by the following link: http://ncov.schanglab.org.cn/download.php?dir=202006011431500000. For the top rank binding modes, the predicted binding energy of Top 1 was -8.3 kcal/mol, and the L_RMSD was 1.97 Å. The output interface is shown as follows.
Main protease and its inhibitors are taken as an example for small molecule batch docking. Main protease is selected as the protein target. The “Batch Docking” mode is selected as the computational type and the mol2 format of its 11 inhibitors are uploaded as the input ligands. Exhaustiveness option is 12.The input interface is shown as follows.
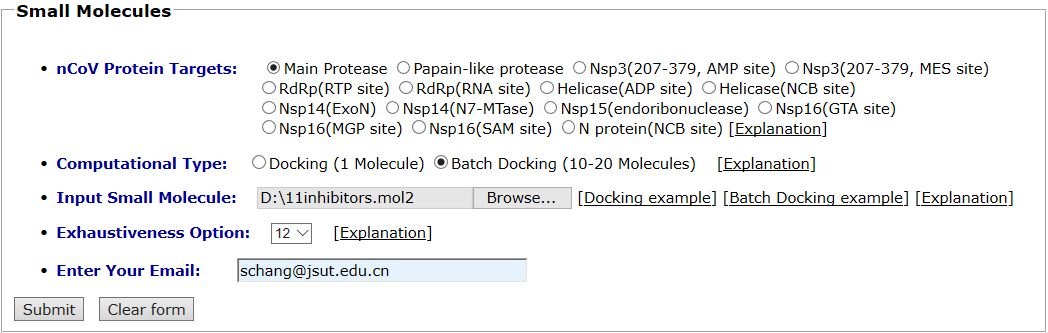
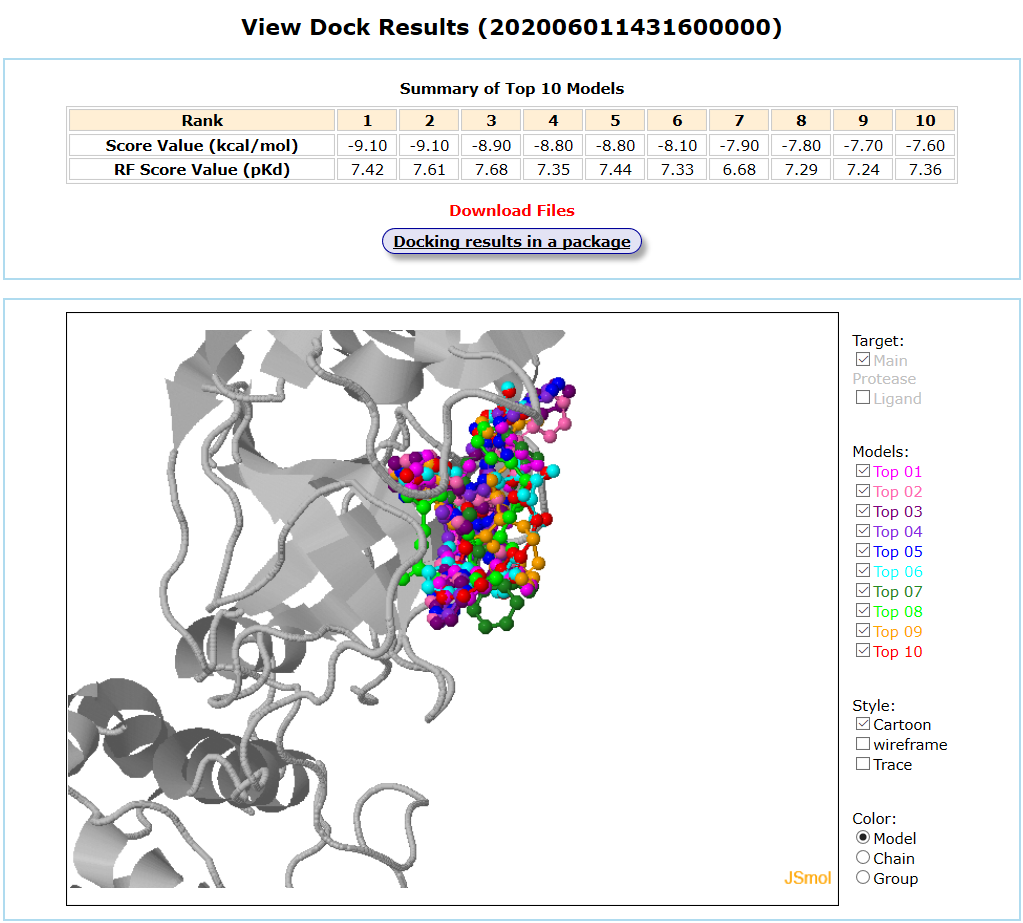
Results are available on the page by the following link: http://ncov.schanglab.org.cn/download.php?dir=202006011431600000. The output interface is shown as follows.
The post-fusion state of S2 segment of spike protein is taken as an example for peptide docking. A 5-helices structure extracted from the post-fusion state of 6-helices S2 is selected as the protein target. The pdb file of ligand peptide was uploaded and the default global docking mode was chosen. The input interface is shown as follows.
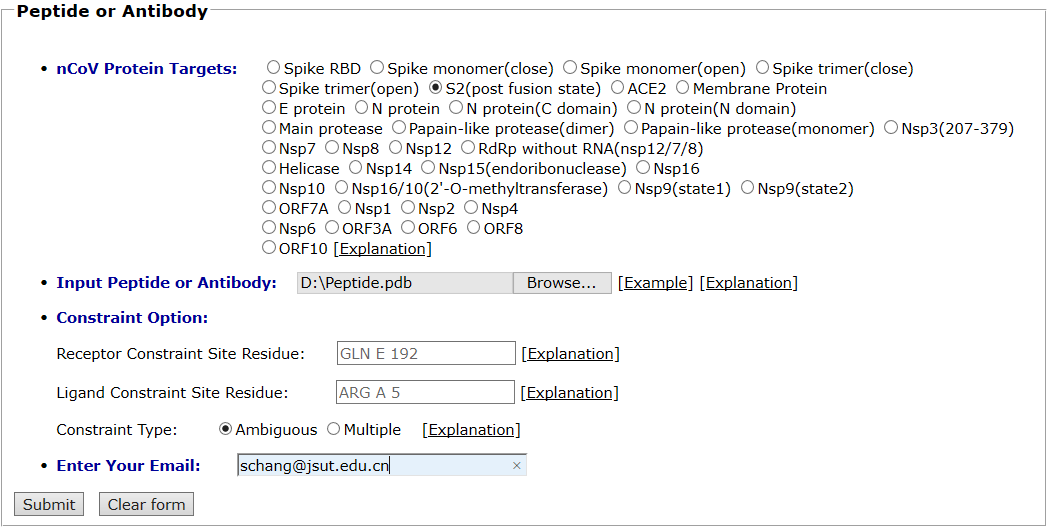
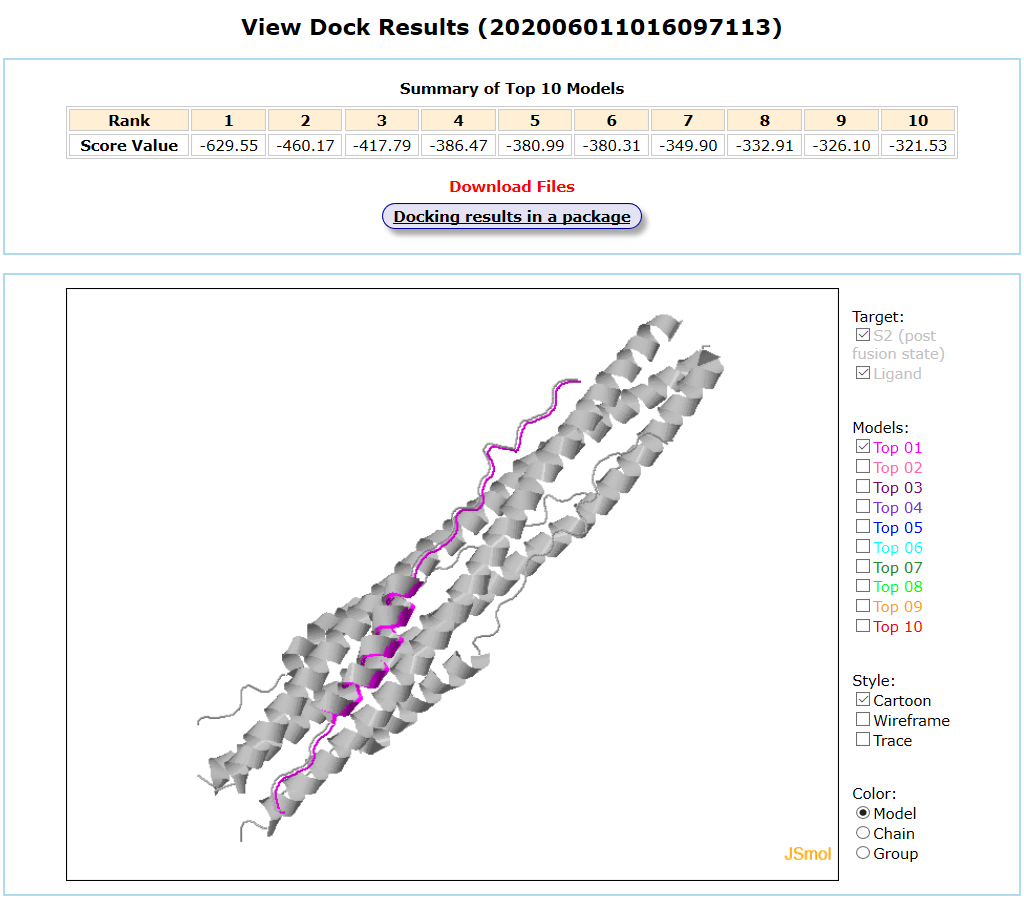
Results are available on the page by the following link: http://ncov.schanglab.org.cn/download.php?dir=202006011016097113. For the top rank binding modes, the scoring value of Top 1 binding mode was -629.6, and the L_RMSD was 0.93 Å. The output interface is shown as follows.
The spike protein and antibody are taken as an example for antibody docking. The spike protein is selected as the protein target. The pdb file of antibody (7BZ5) was uploaded and the default global docking mode was chosen. The input interface is shown as follows.
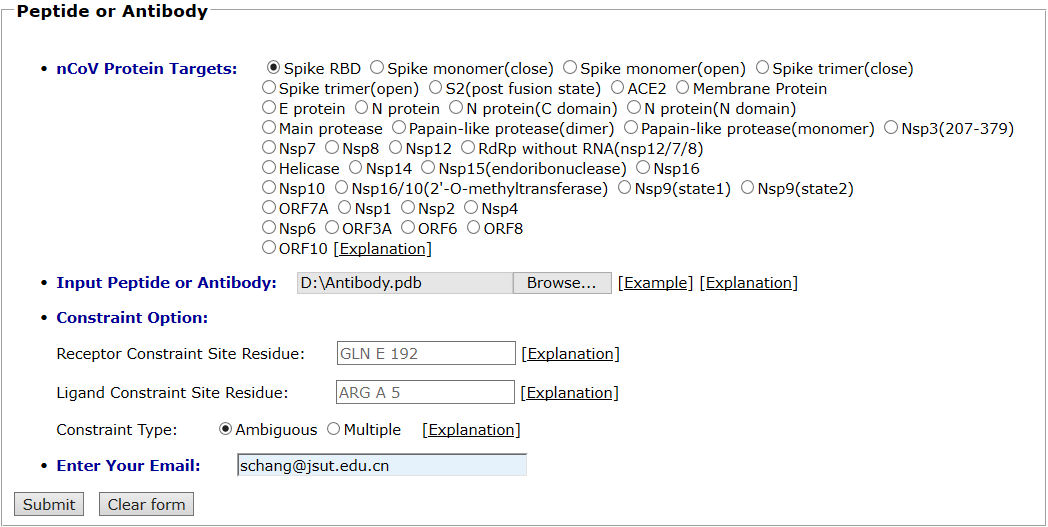
Results are available on the page by the following link: http://ncov.schanglab.org.cn/download.php?dir=202005162147553997. For the top rank binding modes, the scoring value of Top 1 binding mode was -440.9, and the L_RMSD was 1.14 Å. The output interface is shown as follows.
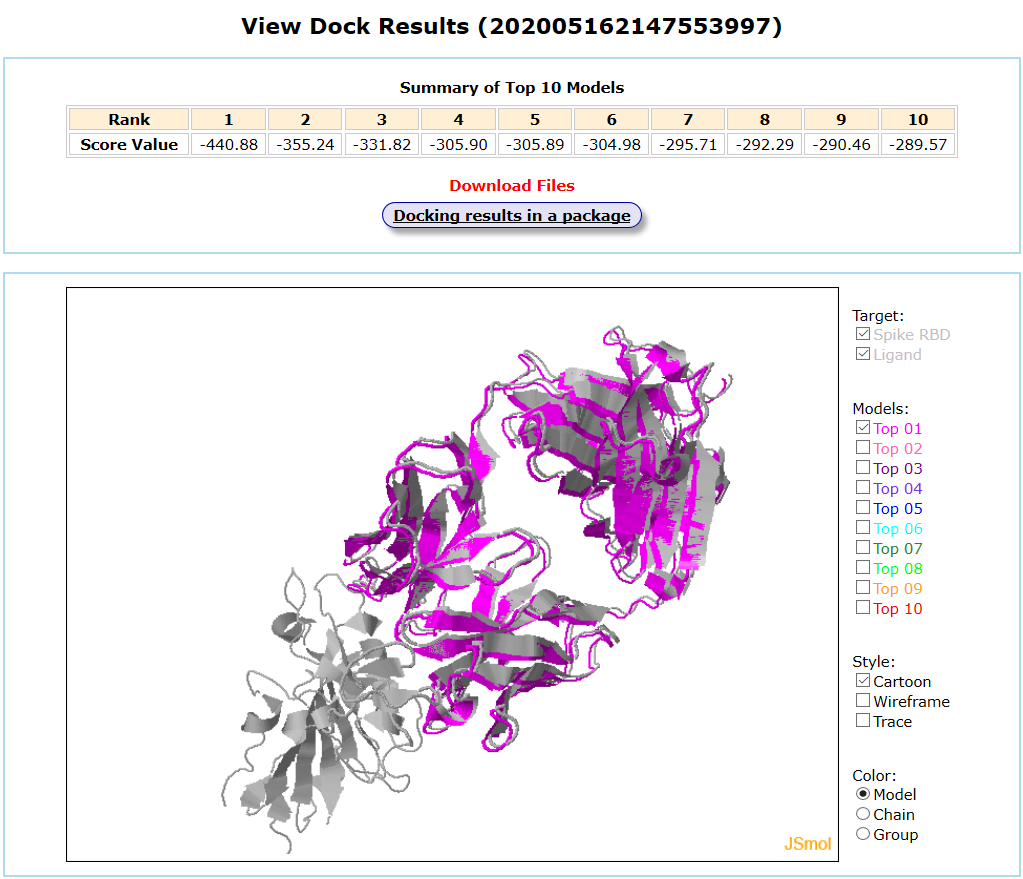
If you have any suggestions or questions, please Contact Us.